Mauricio Cafiero Computational Chemistry, Machine Learning and AI Research
Current Projects
- The Fall 2025 semester saw our participation in two hackathons! For the LLMs in Chemistry and Materials Hackathon
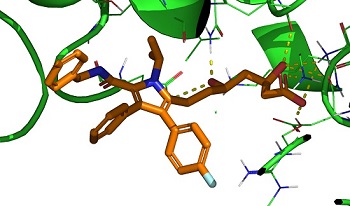
we developed a novel docking code that uses he UMA MLIP to find poses and predict the binding affinity of a ligand to a protein and perform
dynamics on the best pose. See UMADock!
For the Gradio/Huggingface MCP 1st birthday hackathon we created a MOdular DRug design AGent, MoDrAg! She can help with finding
molecular properties, generating libraries of molecules, predicting IC50, and docking.
See MoDrAg!
You can also chat with an OpenAI model using MoDrAg's drug-design sub-agents. See
MoDrAg_Chat!
We also taught a module on Python, Machine Learning, and AI for Chemistry, which included creating MLPs with Pytorch and
creating chatbots with Huggingface models. See the notebooks for the class and other materials at
CafChemTeach!
- Our latest
work in PCCP is the first to show how using a temperature ramp during molecule generation with a GPT
(or any autoregressive model) can make better libraries of molecules.
- We've been making tools for computational chemistry using models like Meta's
UMA MLIP,
Chemeleon and Boltz2. Check out the
CafChem tools for Python here, or the
CafChem HuggingFace spaces/MCP tools here!
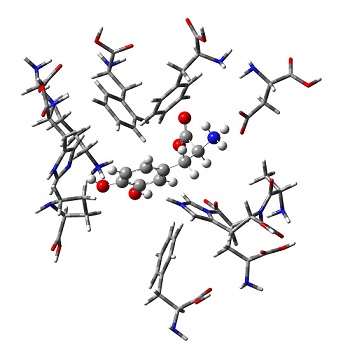
- We recently published a set of CCSD(T)/CBS benchmarks for biologically relevant catecholic systems. These benchmarks
provide accurate information that can aid in drug design for Parkinson's Disease. We also evaluated 21 DFT methods
and found the most accurate functionals for these systems.
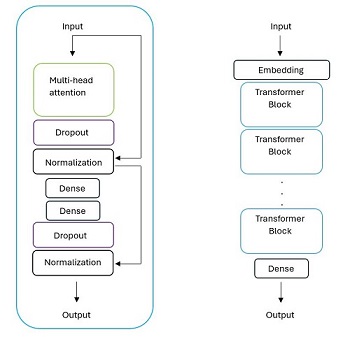
- Designing generative, pre-trained (GPT) models to create virtual screening libraries for drug design.
We have created transformer-based GPTs for creating libraries of molecules to inhibit HMG coenzyme A reductase
(anti-cholesterol drugs, read the paper in
Journal of Chemical Information and Modeling), and are currently working
on RNN and transformer-based GPTs for inhibiting the MAOB enzyme for possible action against Parkinon's Disease.
- See our newest web-apps: upload any dataset with a SMILES column and a target column (values you want to predict) and train
a neural network to predict those target values! Neural netowrks for datasets! Also,
upload or randomly generate a basis set of gaussian orbitals or explicitly correlated gaussian geminals for small atoms and use
stochastic optimization to find the ground state energy! Build a basis set!
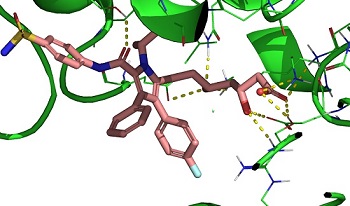
- We recently published a paper on three-body interactions in
protein-ligand binding (where the three bodies are the ligand and two amino-acid residues) and published a
second paper which examines general three-body systems
important to drug design and evaluates many families of DFT methods for their accuracy.
- Much of our work is in DFT-studies of protein-ligand binding, usually in the area of drug-design for Parkinson's Disease.
This recent work describes how paracetamol may have some action in
the relief of symptoms of Parkinson's Disease.
- We are developing the CafChem Javascript-based WebApps for teaching chemistry,
including a introduction to machine-learning
neural networks, where the user can train their own neural network to fit a potential energy surface. We also have apps for
visualizing liquid/solid surface interactions, predicting rovibrational spectra, drawing molecules and predicting their ADME
properties for drug design, and performing calculations for various chemistry practicals.
This is the work of Dr. Mauricio Cafiero and may be used widely though attribution is appreciated.